JMC:金坚教授团队发现首个靶向AKT变构位点的PROTAC
时间:2022-10-30 20:35:39 热度:37.1℃ 作者:网络
AKT是癌症治疗的一个重要靶点。近年来,在开发ATP竞争性的活性位点和变构位点的AKT抑制剂方面取得了重大进展,多款AKT小分子抑制剂获批临床。此外,研究人员也开发了多种基于AKT活性位点的PROTAC,包括MS21。MS21可以有效诱导PI3K/pten通路突变的肿瘤细胞中AKT的降解,并有效抑制肿瘤细胞增殖生长,但在KRAS/BRAF突变细胞中却显得束手无策,无法降解AKT蛋白。
近日,西奈山伊坎医学院金坚教授团队和Ramon E. Parsons团队在知名药物化学期刊JMC上发表题为“Novel Allosteric Inhibitor-Derived AKT Proteolysis Targeting Chimeras (PROTACs) Enable Potent and Selective AKT Degradation in KRAS/BRAF Mutant Cells”的文章。
作者开发了首个基于变构位点的新型AKT PROTAC 62,并且化合物62在KRAS/BRAF突变的癌细胞以及PI3k/pten突变的癌细胞中均显示出强效的AKT降解活性和抗增殖活性。此外,62在通过腹腔内给药的小鼠体内表现出良好的生物利用度和PK性质。
1、AKT靶点介绍
AKT(蛋白激酶b)是一种丝氨酸/苏氨酸激酶,属于丝氨酸蛋白激酶家族。AKT作为人类癌症中最常见的PI3K/AKT/m-TOR信号级联的中心节点,在调节癌症特征中发挥关键作用,如细胞增殖、代谢、转移、侵袭性和肿瘤细胞的存活(图1)。
AKT由三个高度同源的亚型组成,AKT1/2/3,它们都有三个进化上保守的结构域:一个N端PH结构域,一个中心激酶催化结构域和一个C端调控结构域。研究表明,AKT的过度活化和过表达与许多总体预后较差的人类癌症有关。

图1.AKT介导的信号通路
2、AKT小分子抑制剂研究进展
AKT是一种极具临床潜力的药物靶点。在过去数十年来,基于AKT领域的药物研发已经取得了积极的研究成果,多款AKT小分子抑制剂相继获批临床,包括靶向AKT活性位点的小分子抑制剂:GSK690693、GDC-0068、AZD5363和靶向AKT变构位点的抑制剂:MK-2206、ARQ-092、TAS-117。进展最快的是罗氏的GDC-0068和阿斯利康的AZD5363,目前均处于临床三期。
然而,基于活性位点的AKT抑制剂具有严重的副作用,包括高血糖、血小板减少和感染,而AKT变构抑制剂在人体临床试验中也表现出有限的疗效。
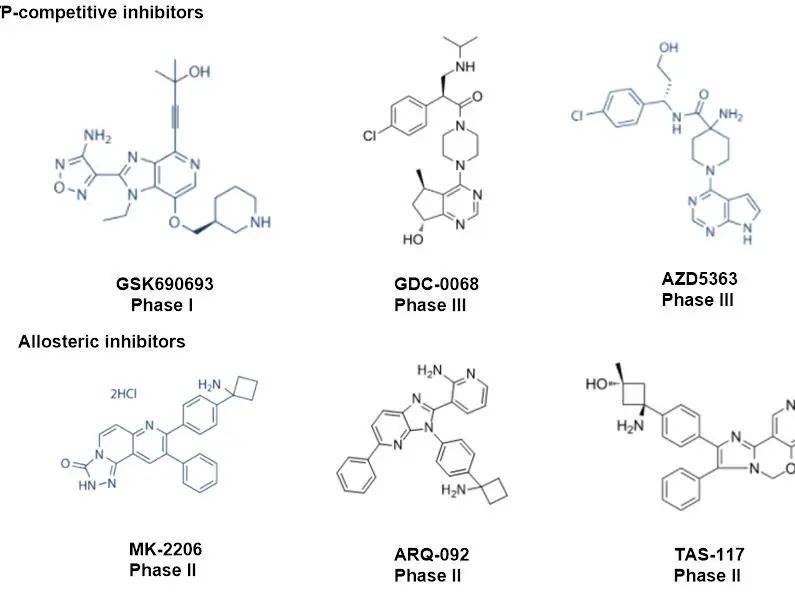
图2.进入临床试验的AKT小分子抑制剂
3、已报道的基于活性位点抑制剂的AKT PROTAC
基于靶向蛋白水解嵌合体PROTAC或分子胶的靶向蛋白降解技术(TPD)是一种通过降解致病靶蛋白来治疗疾病的治疗策略。通常,由PROTAC或分子胶诱导的泛素−蛋白酶体系统(UPS)介导的降解可以同时靶向癌蛋白的催化和非催化结构域,这可能产生比小分子激酶抑制剂更大的疗效。
近年来,研究人员已经开发了多种靶向活性位点的AKT PROTAC(图3),其中,MS21是已报道最有效的AKT降解剂,然而MS21不能有效诱导携带KRAS或BRAF突变的癌细胞中AKT的降解。为了克服MS21等靶向活性位点PROTAC在携带KRAS/BRAF突变的癌细胞较差的降解AKT能力。作者另辟蹊径,试图从AKT的变构抑制剂切入,开发新型的靶向变构位点的AKT降解剂。

图3.已报道的靶向活性位点的AKT PROTAC
下面让我们一起走进这篇文献:
1. 基于ARQ-092 PROTAC的设计思路及合成
研究表明,与ATP竞争性结合的AKT活性位点抑制剂优先结合激活的AKT(磷酸化的AKT,p-AKT)。由于在KRAS/BRAF突变的肿瘤细胞系中,磷酸化AKT蛋白水平较低,这就影响了与ATP竞争性结合的活性位点抑制剂的效果,从而导致耐药。
与靶向活性位点抑制剂不同,AKT变构抑制剂可以结合并稳定AKT的失活形式。因此,靶向AKT变构位点的PROTAC有可能在含有KRAS/BRAF突变的癌细胞中参与结合失活的AKT,这就可能诱导MS21耐药肿瘤细胞系中AKT的降解。
首先,作者选取了AKT变构抑制剂ARQ-092作为AKT PROTAC的靶头化合物,随后通过对ARQ-092与AKT1的晶体复合物进行分析,作者发现ARQ-092中的左侧苯环伸向溶剂区(图4)。因此,作者选用该位点作为连接位点进行衍生化,分别合成了末端是羧基和氨基的化合物1(末端羧基)和2(末端氨基)。随后,作者基于化合物1和2开展PROTAC合成工作。

图4. ARQ-092与AKT1的晶体复合物及化合物1和2的化学结构
VHL配体VHL-1和CRBN配体泊马度胺是最常用的两种E3泛素连接酶配体。作者同时采用这两种E3配体,分别合成了基于VHL配体和CRBN配体两种类型的AKT PROTAC。
最先开展的是基于化合物1(末端羧基)的PROTAC合成(图5)。其中,化合物3-20是基于VHL配体的PROTAC,作者分别合成了不同长度的聚乙二醇和烷烃链两种类型PROTAC。同时作者也合成了基于泊马度胺的PROTAC 21-32,同样采用了不同长度的聚乙二醇和烷烃链两种连接链类型。
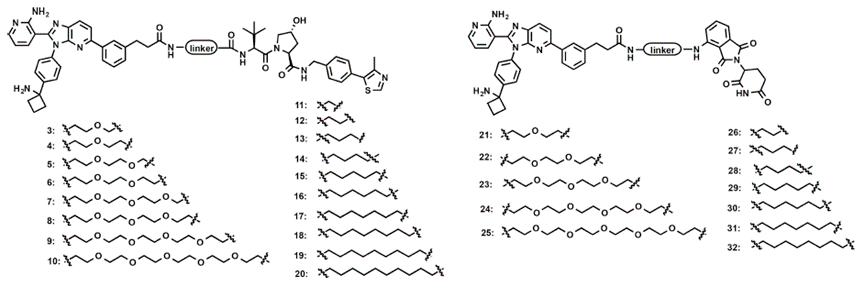
图5.基于化合物1的AKT PROTAC
接下来,作者合成了基于化合物2(末端氨基)PROTAC 33-61(图6)。其中,化合物33-49是基于VHL配体的PROTAC,化合物50-61是基于泊马度胺的PROTAC。并且作者同样也考察了不同长度的聚乙二醇和烷烃链的连接链类型,其中化合物33-41和57-61是聚乙二醇类型的PROTAC,化合物42-49和50-56是烷烃链类型的PROTAC。

图6.基于化合物2的AKT PROTAC
2. PROTAC蛋白降解能力初筛实验
接下来,作者在PC3细胞系(人前列腺癌细胞)上筛选合成的PROTAC 3-61对AKT蛋白的降解能力(图7)。蛋白降解实验表明基于VHL配体的PROTAC比基于泊马度胺的PROTAC具有更强的降解能力,且烷烃链比聚乙二醇链更有效。作者从构效关系研究发现了PROTAC 20、48和49具有较强的AKT降解能力,并可以有效抑制下游信号传导,如:p-akt、p-pras40和p-s6。
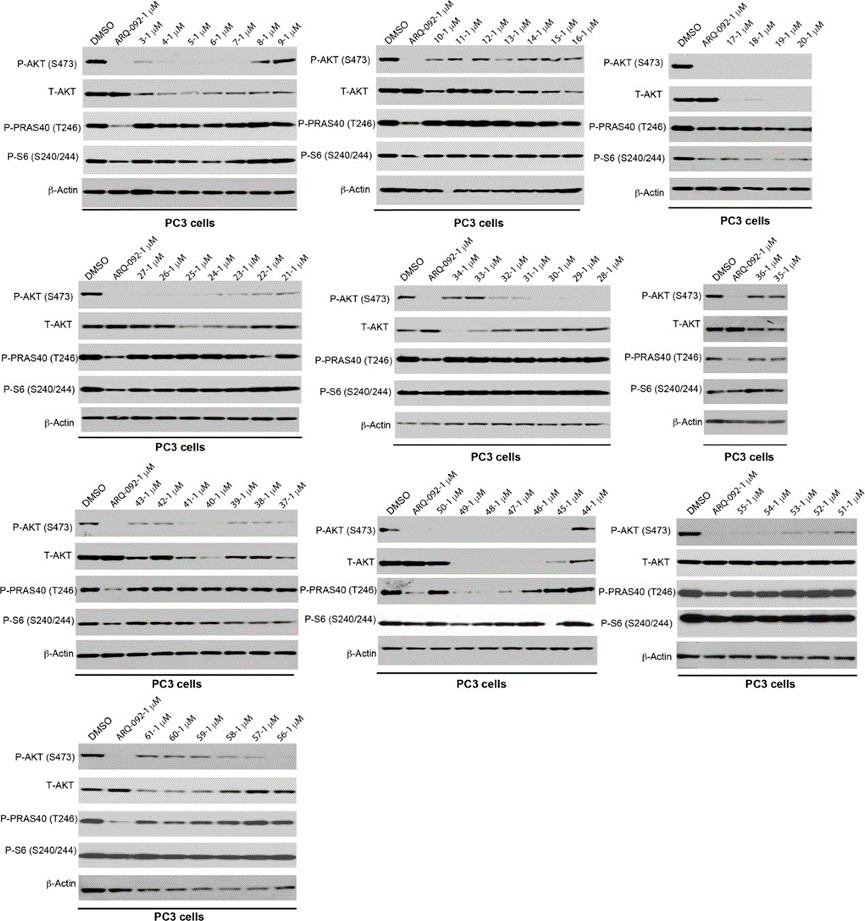
图7. 化合物3-61的蛋白降解实验
3. 基于最优化合物20的进一步结构优化及后续生物活性研究
有研究表明,VHL-2(VHL-1中苄位甲基取代的结构)对VHL具有更高的亲和力。因此,作者选用了降解活性最好的PROTAC 20进行进一步结构改造,将20中的VHL-1替换成具有更高亲和力的VHL-2,得到了PROTAC 62。作者也分别合成了化合物63和64作为阴性对照化合物(图8)。
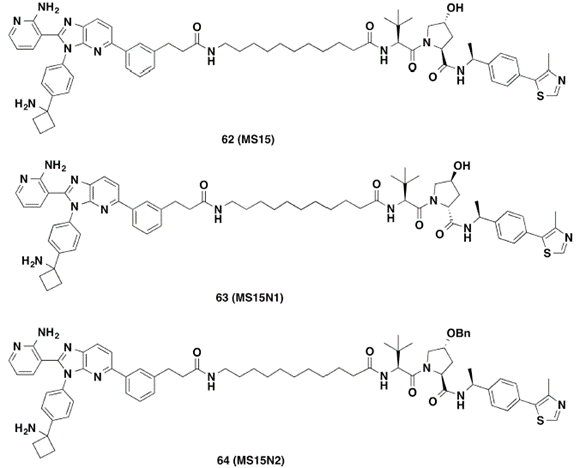
图8. PROTAC 62及阴性对照63和64的化学结构
首先,作者分别测试了PROTAC 62、63、64及靶头化合物ARQ-092对AKT三种不同亚型(AKT1、AKT2、AKT3)的酶抑制活性(图9)。实验结果显示,相较于靶头化合物ARQ-092,PROTAC表现出减弱的抑制活性,但对于AKT1/2/3都具有抑制活性。

图9. PROTAC 62、63、64及ARQ-092的酶活测试
接下来,作者测试了PROTAC 62、63、64在MS21(已报道的降解活性最好的靶向活性位点PROTAC)耐药且KRAS突变的结直肠癌细胞系SW620的降解活性(图10)。实验结果表明,化合物62能够呈现浓度依赖方式有效诱导SW620中的AKT降解,且DC50为23±16 nM,而阴性对照63和64无降解活性。
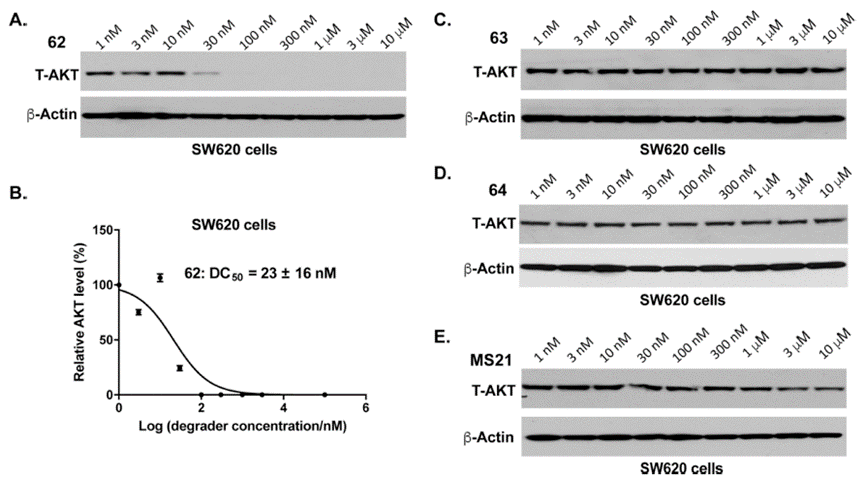
图10.化合物62在sw620中的降解实验
随后,作者比较了62和MS21在其他几种携带KRAS或BRAF突变的癌细胞类型中的AKT降解效应(图11)。蛋白质印迹实验发现,化合物62可以有效诱导各种KRAS或BRAF突变的癌细胞中AKT的降解,而MS21表现出较差的AKT降解能力。

图11. 62和MS21在各种携带KRAS或BRAF突变的癌细胞蛋白降解实验
作者继续在SW620细胞系进行了蛋白降解机制研究(图12),实验结果表明化合物62可以以时间依赖的方式诱导AKT降解,并可以有效抑制磷酸化PRAS40蛋白水平。当加入NAE抑制剂MLN4924或蛋白酶体抑制剂MG132后,可以有效逆转AKT的降解效果。基于上述实验结果,作者认为化合物62诱导AKT蛋白的降解效果依赖于泛素蛋白酶体途径。

图12. 蛋白降解机制研究实验
为了探究PROTAC 62蛋白质组范围内的降解选择性,作者对化合物62进行了基于全局串联质谱标签TMT定量蛋白分析的蛋白质组学研究,实验结果表明化合物62不能降解其他靶点蛋白,具有较好的靶点选择性(图13)。
 图13.化合物62的蛋白质组学研究
图13.化合物62的蛋白质组学研究
接下来,作者在SW620细胞系上测试了化合物62、靶头化合物ARQ-092、阴性对照64及降解剂MS21的抗增殖活性(图14)。从细胞集落形成实验可以看出,化合物62对SW620抑制活性与ARQ-092相当,而阴性对照化合物64和MS21表现出较弱抑制活性。

图14. 化合物62、64、ARQ-092、MS21的细胞集落形成实验
最后,作者对化合物62进行了PK研究(图14),评估化合物62在小鼠体内的生物利用度。实验结果发现在给药后0.5 h可达到最大血药浓度(Cmax = 1 μM),血药浓度保持在100 nM以上至少可维持12小时。这些结果表明,化合物62表现出较好的PK性质,在体内可以获得足够的血浆暴露。

图14. 化合物62在小鼠血浆浓度测试
总结
金坚课题组报道了首个靶向变构位点的AKT PROTAC 62,PRORAC 62能够有效诱导KRAS/BRAF突变肿瘤细胞的AKT降解,这可能为靶向活性位点的MS21耐药KRAS/BRAF突变癌症提供一个潜在的策略。同时,化合物62表现出较强的降解活性、较好的靶点选择性以及较好的PK性质,这是靶向AKT PROTAC领域的一大步,也是靶向AKT领域的药物研发的一大步。
西奈山伊坎医学院金坚教授团队长期深耕于靶向蛋白降解领域,致力于蛋白降解药物的设计与开发,以及推进这些技术的临床应用与转化。该团队已经成功开发了多种靶点的PROTAC,克服了传统小分子药物的种种缺陷,为靶向蛋白降解领域的研发不断注入强心剂。包括:组蛋白甲基转移酶NSD家族NSD2/3,激酶家族AKT和ALK,EGFR以及表观遗传家族靶点EZH2等。
此外,金坚教授团队也开发了其他新型PROTAC技术:
1、叶酸包裹的Folate-Caged PROTACs,该类PROTAC进一步提高了PROTAC的细胞选择性。
2、基于寡核苷酸链的TF-PROTACs,成功实现了对不可成药转录因子的降解。
3、光诱导PROTACopto-PROTAC,实现了PROTAC的可控激活。
4、靶向WDR5 和Ikaros的双靶点PROTAC
5、开发新型的E3连接酶KEAP1配体,并成功用于BRD3/4的降解。进一步拓展了靶向蛋白质降解的有限工具箱。
参考文献
1、Novel Allosteric Inhibitor-Derived AKT Proteolysis TargetingChimeras (PROTACs) Enable Potent and Selective AKT Degradation in KRAS/BRAF Mutant Cells.J Med Chem (IF: 7.45; Q1) . 2022 Oct 27;65(20):14237-14260.
2、Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discovery 2005, 4 (12), 988−1004.
3、Harnessing the E3 Ligase KEAP1 for Targeted Protein Degradation. DOI: 10.1021/jacs.1c04841. J. Am. Chem. Soc.2021, 143, 37, 15073–15083
4、4、TF-PROTACs Enable Targeted Degradation of Transcription Factors.J. Am. Chem. Soc.2021, 143, 23, 8902–8910. doi.org/10.1021/jacs.1c03852

